

| |
zurück | |
| Methoden zur Bestimmung der physikalisch-chemischen Eigenschaften
A.4. Dampfdruck |
Anhang V zur RL 67/548/EWG |
A.4. 1. Methoden
Den meisten der hier beschriebenen Methoden liegt die OECD-Prüfrichtlinie (1) zugrunde. Die Grundprinzipien sind in (2) und (3) angegeben.
A.4. 1.1. Einleitung
Zur Durchführung der Prüfung ist es nützlich, Vorinformationen über die Struktur, die Schmelz- und die Siedetemperatur der Prüfsubstanz zu haben.
Es gibt keine Prüfmethode, die für den gesamten Dampfdruck-Meßbereich geeignet ist. Daher werden zur Messung der Dampfdrucke von 1< 10-4 bis 105 Pa mehrere Methoden empfohlen.
Der Dampfdruck wird gewöhnlich von Verunreinigungen beeinflußt. Der Einfluß hängt in hohem Maße von der Art der Verunreinigung ab.
Wenn in der Probe flüchtige Verunreinigungen vorliegen, die das Ergebnis beeinträchtigen könnten, kann die Prüfsubstanz gereinigt werden. Es kann auch von Nutzen sein, den Dampfdruck für den technischen Stoff anzugeben.
Einige der hier beschriebenen Methoden verwenden eine Apparatur mit Metallteilen. Dies ist bei der Prüfung korrosiver Substanzen zu berücksichtigen.
A.4. 1.2. Definitionen und Einheiten
Der Dampfdruck einer Substanz ist definiert als der Sättigungsdruck über einer festen oder flüssigen Substanz. Im thermodynamischen Gleichgewicht ist der Dampfdruck einer reinen Substanz ausschließlich eine Funktion der Temperatur.
Die zu verwendende SI-Einheit für den Druck ist Pascal (Pa).
Nachstehend einige der früher gebräuchlichen Einheiten mit den entsprechenden Umrechnungsfaktoren:
| 1 Torr (= 1 mm Hg) | = 1,333 ξ 102 Pa |
| 1 Atmosphäre | = 1,013 ξ 105 Pa |
| 1 bar | = 105 Pa. |
Die SI-Einheit für die Temperatur ist Kelvin (K).
Die universelle molare Gaskonstante R beträgt 8,314 J mol-1 K-1
Die Temperaturabhängigkeit des Dampfdrucks wird durch die Clausius-Clapeyron-Gleichung beschrieben:
 + C
+ C
Darin bedeuten:
| P | : der Dampfdruck des Stoffes in Pascal |
| ΔHv | : seine Verdampfungswärme in j mol-1 |
| R | : die universelle molare Gaskonstante in J mol-1 K-1 |
| T | : die thermodynamische Temperatur in K |
| C | : weitere stoffspezifische Konstante. |
A.4. 1.3. Referenzsubstanzen
Referenzsubstanzen müssen nicht in allen Fällen verwendet werden, in denen eine neue Prüfsubstanz untersucht wird. Die Referenzsubstanzen sollten in erster Linie dazu dienen, die Methode von Zeit zu Zeit zu überprüfen und einen Vergleich mit den Ergebnissen aus anderen Methoden zu ermöglichen.
A.4. 1.4. Prinzip der Methoden
Zur Bestimmung des Dampfdrucks werden sieben Methoden vorgeschlagen, die in unterschiedlichen Dampfdruckbereichen eingesetzt werden können. Mit jeder Methode wird der Dampfdruck bei verschiedenen Temperaturen bestimmt. In einem begrenzten Temperaturbereich ist der Logarithmus des Dampfdrucks einer reinen Substanz der Temperatur umgekehrt proportional.
A.4. 1.4.1. Dynamische Methode
Bei der dynamischen Methode wird die Siedetemperatur der Prüfsubstanz bei einem bestimmten vorgegebenen Druck gemessen.
Empfohlener Bereich: 103 bis 105 Pa.
Diese Methode wird ebenfalls für die Bestimmung der Normal-Siedetemperatur empfohlen, wobei sie sich bis zu Temperaturen von 600 K eignet.
A.4. 1.4.2 Statische Methode
Bei diesem Verfahren wird derjenige Dampfdruck gemessen, der sich im thermodynamischen Gleichgewicht im geschlossenen System bei einer gegebenen Temperatur über einer Substanz einstellt. Diese Methode eignet sich für Ein- und Mehrkomponentensysteme (Feststoffe und Flüssigkeiten).
Empfohlener Bereich: 10 bis 105 Pa; unter entsprechenden Vorsichtsmaßnahmen auch von 1 bis 10 Pa.
A.4. 1.4.3 Isoteniskop
Diese genormte Methode beruht ebenfalls auf einem statischen Verfahren, eignet sich jedoch im allgemeinen nicht für Mehrkomponentensysteme. Weitere Informationen können der ASIM-Methode D-2879-86 entnommen werden.
Empfohlener Bereich: von 100 bis 105 Pa.
A.4. 1.4.4 Effusionsmethode: Dampfdruckwaage
Unter Vakuumbedingungen bestimmt man die Substanzmenge, die eine Meßzelle pro Zeiteinheit durch eine Öffnung bekannter Größe so verläßt, daß eine Rückkehr der Substanz in die Meßzelle vernachlässigt werden kann (z.B. durch Messung des Impulses eines Dampfstrahls auf eine empfindliche Waage oder durch Bestimmung des Gewichtsverlusts).
Empfohlener Bereich: 10-3 bis 1 Pa
A.4. 1.4.5. Effusionsmethode: durch Masseverlust oder über eine Kühlfalle
Unter Hochvakuumbedingungen bestimmt man die Masse der Prüfsubstanz, die eine Knudsen-Zelle (4) pro Zeiteinheit in Form von Dampf durch eine Mikroöffnung verläßt, Die ausgeströmte Dampfmasse läßt sich entweder durch Bestimmung des Masseverlusts der Zelle oder durch Kondensation des Dampfes bei niedrigen Temperaturen und chromatographische Bestimmung der verdampften Substanzmenge ermitteln. Der Dampfdruck wird durch Anwendung der Hertz-Knudsen-Formel berechnet.
Empfohlener Bereich: 10-3 bis 1 Pa.
A.4. 1.4.6. Gassättigungsmethode
Ein Strom eines Inertgases wird so über die Prüfsubstanz geleitet, daß er sich mit deren Dampf sättigt. Die Substanzmenge, die von einer bekannten Menge an Trägergas transportiert wird, läßt sich entweder durch Sammeln in einer geeigneten Falle oder durch ein online Analysenverfahren messen. Diese wird dann zur Berechnung des Dampfdrucks bei einer bestimmten Temperatur verwendet.
Empfohlener Bereich: 10-4 bis 1 Pa; unter entsprechenden Vorsichtsmaßnahmen auch von 1 bis 10 Pa.
A.4. 1.4.7. Rotormethode
Das eigentliche Meßelement bei der Rotormethode ist eine kleine Stahlkugel, die sich mit hoher Geschwindigkeit in einem Magnetfeld dreht. Der Gasdruck wird von der druckabhängigen zeitlichen Abnahme der Rotationsgeschwindigkeit der Stahlkugel abgeleitet.
Empfohlener Bereich: 10-4 bis 0,5 Pa.
A.4. 1.5. Qualitätskriterien
In der nachstehenden Tabelle findet sich ein Vergleich der verschiedenen Dampfdruck-Bestimmungsmethoden hinsichtlich ihrer Anwendung, Wiederholbarkeit, Reproduzierbarkeit, ihres Meßbereich sowie existierender Normen.
Tabelle: Qualitätskriterien
| Meßmethode | Substanzen | Geschätzte Wiederhol- barkeit |
Geschätzte Reproduzier- barkeit |
Empfohlener Bereich |
Existierende Norm | |
| Feststoffe | Flüssigkeit | |||||
| 1.4.1. Dynamische Methode | niedr.- schmelz | ja | bis 25 % | bis 25 % | 103 Pa bis 2 x 103 Pa | - |
| 1 bis 5 % | 1 bis 5 % | 2 x 103 Pa bis 105 Pa |
- | |||
| 1.4.2. Statische Methode | ja | ja | 5 bis 10 % | 5 bis 10 % | 10 Pa bis 105 Pa (2) |
NFT 20-048 (5) |
| 1.4.3. Isoteniskop | ja | ja | 5 bis 10 % | 5 bis 10 % | 102 Pa bis 105 Pa |
ASTM-D 2879-86 |
| 1.4.4. Effusionsmethode Dampfdruckwaage | ja | ja | 5 bis 20 % | bis 50 % | 10-3 Pa bis 1 Pa | NFT 20-047(6) |
| 1.4.5. Effusionsmethode Masseverlust | ja | ja | 10 bis 30 % | - | 10-3 Pa bis1 Pa | - |
| 1.4.6. Gassättigungsmethode | ja | ja | 10 bis 30 % | bis 50 % | 10-4 Pa bis 1 Pa (2) | - |
| 1.4.7. Rotormethode | ja | ja | 10 bis 20 % | - | 10-4 Pa bis 0,5 Pa | - |
| (1) abhängig vom Reinheitsgrad der Prüfsubstanz (2) Diese Methoden können unter entsprechenden Vorsichtsmaßnahmen auch im Bereich 1 bis 10 Pa angewendet werden. |
||||||
A.4. 1.6. Beschreibung der Methoden
A.4. 1.6.1. Dynamische Methode
A.4. 1.6.1.1. Apparatur
Die Apparatur besteht im allgemeinen aus einem Siedegefäß mit Aufsatzkühler aus Glas oder Metall (Abbildung 1) sowie einer entsprechenden Einrichtung zum Messen der Temperatur sowie zum Regeln und Messen des Drucks. Die in der Abbildung dargestellte typische Apparatur besteht aus hitzebeständigem Glas und setzt sich aus fünf Teilen zusammen:
Großes, teilweise doppelwandiges Rohr, bestehend aus einer Schliffverbindung, einem Kühler, einem Kühlkolben und einem Einlaß.
Glaszylinder mit Cottrellpumpe, der im Siedebereich des Rohres angebracht ist und innen eine aufgerauhte Oberfläche aus gesintertem Glas besitzt, um Siedeverzüge zu vermeiden.
Die Temperatur wird mit einem geeigneten Temperaturfühler (z.B. Widerstandsthermometer, Mantelthermoelement) gemessen, der durch einen geeigneten Einlaß (z.B. Kernschliffverbindung) in die Apparatur eingetaucht wird und bis an die Meßstelle (Nr. 5, Abbildung 1) reicht.
Die notwendigen Verbindungen zur Druckregel- und Meßeinrichtung werden hergestellt.
Der Rundkolben, der als Puffervolumen dient, wird mittels einer Kapillare mit der Meßapparatur verbunden.
Das Siedegefäß wird durch ein Heizelement (z.B. Heizpatrone) erhitzt, welches von unten in die Glasapparatur eingeführt wird. Der erforderliche Heizstrom wird mit einem Thermoelement eingestellt und geregelt.
Das erforderliche Vakuum zwischen 102 Pa und etwa 105 Pa wird mit einer Vakuumpumpe erzeugt. Ein geeignetes Ventil wird zur Dosierung von Luft oder Stickstoff zwecks Druckregelung (Meßbereich etwa 102 Pa bis 105 Pa) und Belüftung verwendet.
Zur Druckmessung dient ein Manometer.
A.4. 1.6.1.2. Meßvorgang
Zur Bestimmung des Dampfdrucks der Probe mißt man deren Siedetemperatur bei verschiedenen festgelegten Drücken zwischen ungefähr 103 und 105 Pa. Die Siedetemperatur ist erreicht, wenn die Temperatur bei konstantem Druck einen zeitlich konstanten Wert erreicht hat. Diese Methode eignet sich nicht zur Messung der Dampfdrücke schäumender Substanzen.
Die Prüfsubstanz wird in das gereinigte und getrocknete Probengefäß gegeben. Dabei kann es bei nicht pulverförmigen Feststoffen Probleme geben, doch lassen sich diese mitunter durch Erwärmen des Kühlmantels umgehen. Nach dem Einfüllen wird die Apparatur zugeflanscht und die Substanz entgast. Danach stellt man den niedrigsten gewünschten Druck ein und schaltet die Heizung an. Gleichzeitig schließt man den Temperaturfühler an einen Schreiber an.
Das Gleichgewicht ist dann erreicht, wenn bei konstantem Druck eine konstante Siedetemperatur erreicht wird. Besondere Vorkehrungen sind zu treffen, um Siedeverzüge zu vermeiden. Darüber hinaus muß es am Kühler zu einer vollständigen Kondensation kommen. Bei der Bestimmung des Dampfdrucks von niedrigschmelzenden Feststoffen sind Vorkehrungen zu treffen, um eine Blockierung des Kühlers zu vermeiden.
Nach Registrierung des Gleichgewichtspunktes wird ein höherer Druck eingestellt. Dann fährt man auf diese Weise fort, bis ein Druck von 105 Pa erreicht ist (ungefähr 5 bis 10 Messungen insgesamt). Zur Überprüfung müssen die Gleichgewichtsbestimmungen bei abnehmendem Druck wiederholt werden.
A.4. 1.6.2. Statische Methode
A.4. 1.6.2.1. Apparatur
Die Apparatur umfaßt ein Behältnis für die Probe sowie ein Heiz- und Kühlsystem zur Temperierung der Probe sowie eine Vorrichtung zur Messung der Temperatur. Außerdem enthält die Apparatur Instrumente zur Einstellung und Messung des Drucks. Die in Anwendung kommenden Grundprinzipien sind in den Abbildungen 2a und 2b dargestellt.
Der Probenraum (Abbildung 2a) wird auf der einen Seite durch ein geeignetes Hochvakuumventil begrenzt. Auf der anderen Seite ist ein U-Rohr angebracht, das eine geeignete Manometerflüssigkeit enthält. Ein Abzweig des U-Rohres führt zur Vakuumpumpe, zum Stickstoffzylinder oder Belüftungsventil und zu einem Manometer.
Anstelle eines U-Rohres kann ein Manometer mit einer Manometerflüssigkeit (Abbildung 2b) verwendet werden.
Zur Temperierung der Probe bringt man den Probenbehälter zusammen mit dem Ventil und dem U-Rohr oder Druckmesser in ein Bad, das konstant auf ± 0,2 K zu temperieren ist. Die Temperaturmessungen werden an der Außenwand des Probengefäßes oder im Gefäß selbst vorgenommen.
Die Apparatur wird mit einer Vakuumpumpe mit einer vorgeschalteten Kühlfalle evakuiert.
Bei Methode 2a mißt man den Dampfdruck der Substanz indirekt über eine Nullanzeige. Dabei wird berücksichtigt, daß sich die Dichte der Flüssigkeit im U-Rohr durch größere Temperaturschwankungen ändert.
Zur Verwendung als Manometerflüssigkeit lassen sich je nach Druckbereich und chemischem Verhalten der Prüfsubstanz folgende Flüssigkeiten verwenden: Silikonöle, Phthalate. Die Prüfsubstanz darf sich in der Manometerflüssigkeit nicht merklich lösen, noch mit ihr reagieren.
Als Manometerflüssigkeit läßt sich Quecksilber im Bereich von normalem Luftdruck bis 102 Pa, Silikonöle und Phthalate lassen sich auch unter 102 Pa bis hinab zu 10 Pa verwenden. Heizbare Membrankapazitätsmanometer lassen sich sogar unter 10-1 Pa einsetzen. Daneben gibt es noch andere Manometer, die unter 102 Pa verwendet werden können.
A.4. 1.6.2.2. Meßvorgang
Vor der Messung müssen alle Bestandteile der Apparatur von Abbildung 2 gründlich gereinigt und getrocknet werden.
Bei der Methode 2a wird das U-Rohr mit der gewählten Flüssigkeit gefüllt, die bei erhöhter Temperatur entgast werden muß, bevor die Ablesungen vorgenommen werden.
Die Prüfsubstanz wird in die Apparatur gegeben, die dann verschlossen und auf eine zum Entgasen hinreichend tiefe Temperatur gebracht wird. Die Temperatur muß deshalb niedrig genug sein, um sicherzustellen, daß die Luft tatsächlich abgesaugt wird; dennoch darf - bei einem Mehrkomponentensystem - die Zusammensetzung des Stoffes nicht verändert werden. Soweit erforderlich, kann das Gleichgewicht schneller durch Rühren hergestellt werden.
Die Unterkühlung der Probe kann z.B. mit flüssigem Stickstoff (Achtung: Kondensation der Luft, Pumpenflüssigkeit) oder einer Mischung aus Ethanol und Trockeneis vorgenommen werden. Bei Niedrigtemperatur-Messungen kann ein an einen Ultrakryostaten angeschlossenes temperiertes Bad verwendet werden.
Bei geöffnetem Ventil über dem Probenbehälter wird mehrere Minuten lang die Luft abgesaugt. Danach wird das Ventil geschlossen und die Probe auf die niedrigste gewünschte Temperatur abgekühlt. Falls notwendig, ist der Entgasungsvorgang mehrere Male zu wiederholen.
Bei Erhitzung der Probe nimmt der Dampfdruck zu. Dadurch werden die beiden Niveaus der Flüssigkeit im U-Rohr verändert. Zum Ausgleich wird Stickstoff oder Luft über ein Ventil in die Apparatur geleitet, bis die Niveaus der Manometerflüssigkeit erneut den Gleichstand erreicht haben. Der dafür erforderliche Druck läßt sich mit einem Präzisionsmanometer bei Raumtemperatur ablesen. Dieser Druck entspricht dem Dampfdruck der Substanz bei dieser Meßtemperatur.
Methode 2b ähnelt dem hier beschriebenen Verfahren, doch wird der Dampfdruck direkt abgelesen.
Die Temperaturabhängigkeit des Dampfdrucks wird in genügend kleinen Intervallen (insgesamt 5 bis 10 Meßpunkte) bis zum gewünschten Maximum bestimmt. Bei niedrigen Temperaturen vorgenommene Ablesungen sind zwecks Überprüfung zu wiederholen.
Wenn die aus den wiederholten Ablesungen stammenden Werte nicht mit der bei steigender Temperatur erhaltenen Kurve übereinstimmen, kann dies eine der nachstehend genannten Ursachen haben:
A.4. 1.6.3. Isoteniskop
Für eine vollständige Beschreibung dieser Methode wird auf (7) verwiesen. Das Prinzip des Meßgeräts zeigt Abbildung 3. Ebenso wie die in 1.6.2. beschriebene statische Methode eignet sich das Isoteniskop zur Untersuchung von Feststoffen und Flüssigkeiten.
Bei der Untersuchung von Flüssigkeiten verwendet man diese gleichzeitig als Anzeigesäule im Hilfsmanometer. Eine Flüssigkeitsmenge, ausreichend, um den Boden und den kurzen Schenkel des Manometers zu füllen, wird in das Isoteniskop gefüllt. Danach wird dieses an ein Vakuumsystem angeschlossen, evakuiert und schließlich mit Stickstoff gefüllt. Evakuierung und Reinigung des Systems werden zweimal wiederholt, um den restlichen Sauerstoff zu entfernen. Das gefüllte Isoteniskop wird in eine horizontale Lage gebracht, so daß sich die Prüfsubstanz als dünne Schicht im Bodenteil und im Manometer (U-Rohr) verteilt. Danach wird der Druck des Systems auf 133 Pa reduziert und die Probe vorsichtig erwärmt, bis sie eben zu sieden anfängt (Entfernung aufgelöster Gase). Anschließend wird das Isoteniskop in eine solche Lage gebracht, daß die Probe in den unteren Teil des U-Rohres und den kurzen Schenkel des Manometers zurückfließt, so daß beide vollständig mit Flüssigkeit gefüllt sind. Der Druck wird wie beim Entgasen konstant gehalten und die ausgezogene Plättchen unter kleiner Flamme erhitzt, bis sich der Dampf der Prüfsubstanz ausreichend ausdehnt, um einen Teil der Substanz aus dem oberen Teil des U-Rohres und dem Manometerschenkel in den Manometer-Teil des Isoteniskops zu verdrängen und einen mit Dampf gefüllten stickstoffreien Raum zu schaffen.
Danach wird das Isoteniskop in ein Bad mit konstanter Temperatur gegeben und der Druck des Stickstoffs an den Druck der Prüfsubstanz angeglichen. Der Gleichstand beider Drücke wird vom Manometer-Teil des Isoteniskops angezeigt. Im abgeglichenen Zustand sind der Dampfdruck des Stickstoffs und der Dampfdruck der Prüfsubstanz gleich.
Bei der Untersuchung von Feststoffen werden je nach Druck- und Temperaturbereich die in 1.6.2.1 aufgeführten Manometerflüssigkeiten benutzt. Die entgaste Manometerflüssigkeit wird in eine Ausbuchtung am langen Schenkel des Isotensikops gefüllt. Dann wird der zu prüfende Feststoff in das Bodenteil eingebracht und bei erhöhter Temperatur entgast. Danach wird das Isoteniskop geneigt, damit die Manometerflüssigkeit in das U-Rohr fließen kann. Zur Messung des Dampfdrucks in Abhängigkeit von der Temperatur verfährt man wie in 1.6.2. angegeben.
A.4. 1.6.4. Effusionsmethode: Dampfdruckwaage
A.4. 1.6.4.1. Apparatur
In der Literatur werden verschiedene Ausführungen der Apparatur beschrieben (1). Die hier beschriebene Apparatur dient zur Darstellung des allgemeinen Funktionsprinzips (Abbildung 4). In Abbildung 4 sind die Hauptbestandteile des Gerätes wiedergegeben: ein Hochvakuum-Behälter aus Edelstahl oder Glas, Ausrüstungen zur Erzeugung und Messung eines Vakuums sowie eingebaute Bauteile zur Messung des Dampfdrucks auf einer Waage. Die Apparatur schließt folgende Einbauten ein:
A.4. 1.6.4.2. Meßvorgang
Man füllt den Behälter mit der Prüfsubstanz und schließt ihn mit dem Deckel. Die Blende mit dem Kühlkasten wird über den Ofen geschoben. Dann wird die Apparatur geschlossen, und die Vakuumpumpen werden eingeschaltet. Vor Beginn der Messungen sollte der Enddruck ungefähr 10-4 Pa betragen. Ab 10-2 Pa beginnt man mit dem Kühlen des Kühlkastens.
Nach Erreichen des erforderlichen Vakuums beginnt man mit der Kalibrierungsreihe bei der niedrigsten gewünschten Temperatur. Man stellt die entsprechende Öffnung im Deckel ein; der Dampfstrahl passiert die direkt darüber befindliche Blende und trifft auf die gekühlte Waagschale. Die Waagschale muß groß genug sein, damit der gesamte durch die Blende geführte Strahl auf sie auftrifft. Der Impuls des Dampfstrahls übt eine Kraft auf die Waagschale aus, und die Moleküle kondensieren auf ihrer gekühlten Oberfläche.
Durch diesen Impuls und die gleichzeitige Kondensation wird ein Signal auf dem Registriergerät erzeugt. Die Auswertung der Signale ergibt zwei Informationen:

Dabei bedeuten:
| G = | Verdampfungsgeschwindigkeit (kg s-1 m-2) |
| M = | Molekulargewicht (g mol-1) |
| T = | Temperatur (K) |
| R = | universelle molare Gaskonstante (J mol-1 K-1) |
| p = | Dampfdruck (Pa). |
Nach Erreichen des erforderlichen Vakuums beginnt man die Meßreihe bei der niedrigsten gewünschten Temperatur.
Im weiteren Verlauf der Messung steigert man die Temperatur in kleinen Intervallen, bis der höchste gewünschte Temperaturwert erreicht ist. Anschließend wird die Probe wieder abgekühlt, und man kann gegebenenfalls eine zweite Dampfdruckkurve aufzeichnen. Falls der zweite Durchgang die Resultate des ersten nicht bestätigt, ist dies unter Umständen darauf zurückzuführen, daß sich die Substanz im untersuchten Temperaturbereich zersetzt.
A.4. 1.6.5. Effusionsmethode - durch Masseverlust
A.4. 1.6.5.1. Apparatur
Die verwendete Apparatur besteht aus den folgenden Hauptbestandteilen:
In Abbildung 5 ist als Beispiel ein elektrisch beheizter Aluminiumbehälter mit 4 Effusionszellen aus Edelstahl dargestellt. Die Edelstahlblende (etwa 0,3 mm dick) hat eine Effusionsöffnung von 0,2 bis 1,0 mm Durchmesser und wird mit der Effusionszelle über einen Deckel mit Gewinde verbunden.
A.4. 1.6.5.2. Meßvorgang
Referenz- und Prüfsubstanz werden in jede Effusionszelle gefüllt, die Metallblende mit Hilfe des Gewindedeckels gesichert und jede Zelle auf 0,1 mg genau gewogen. Danach wird die Zelle in die temperierte Apparatur gegeben, die schließlich bis auf weniger als ein Zehntel des erwarteten Drucks evakuiert wird. Dann wird die Apparatur in definierten Zeitabständen zwischen 5 und 30 Stunden belüftet und der Masseverlust der Effusionszelle durch erneutes Wiegen bestimmt.
Um sicherzustellen, daß die Ergebnisse nicht durch flüchtige Verunreinigungen beeinflußt werden, wird die Zelle in definierten Zeitabständen erneut gewogen. Dadurch soll geprüft werden, ob die Verdampfungsgeschwindigkeit mindestens über zwei Zeitabstände konstant bleibt.
Der Dampfdruck p in der Effusionszelle wird errechnet durch

Dabei bedeuten
| p | = Dampfdruck (Pa) |
| m | = Masse der Substanz, die im Verlauf der Zeit t aus der Zelle ausströmt (kg) |
| t | = Zeit (s) |
| A | = Fläche des Loches (m2) |
| K | = Korrekturfaktor |
| R | = universelle Gaskonstante (J mol-1 K-1) |
| T | = Temperatur (K) |
| M | = Molekulargewicht (kg mol-1) |
Der Korrekturfaktor K hängt vom Verhältnis Länge/Radius der zylindrischen Öffnung ab:
| Verhältnis: | 0,1 | 0,2 | 0,6 | 1,0 | 2,0 |
| K: | 0,952 | 0,909 | 0,771 | 0,672 | 0,514 |
Die obige Gleichung kann dann wie folgt geschrieben werden:

Dabei ist die Konstante der Effusionszelle.
die Konstante der Effusionszelle.
Die Konstante E der Effusionszelle läßt sich mit Hilfe folgender Gleichung mittels Referenzsubstanzen bestimmen (2,9):

Dabei sind
p(r) = der Dampfdruck der Referenzsubstanz (Pa)
M(r) = das Molekulargewicht der Referenzsubstanz (kg/mol).
A.4. 1.6.6. Gassättigungsmethode
A.4. 1.6.6.1. Apparatur
Eine für diesen Test verwendete typische Apparatur besteht aus einer Reihe von in Abbildung 6a dargestellten und nachstehend beschriebenen Bestandteilen (1).
Trägergas:
Das Trägergas darf mit der Prüfsubstanz nicht chemisch reagieren. Gewöhnlich ist Stickstoff als Trägergas geeignet,1doch zuweilen kann die Verwendung anderer Gase erforderlich sein (10). Das verwendete Gas muß trocken sein (siehe Abbildung 6a, Ziffer 4: Sensor zur Messung der relativen Feuchtigkeit).
Durchflußkontrolle:
Ein geeignetes Regelsystem zur Kontrolle des Gasstromes ist notwendig, um einen konstanten und wahlweise einstellbaren Gasfluß durch die Sättigungssäule zu gewährleisten.
Kühlfallen zum Niederschlagen des Dampfes:
Ihre Wahl hängt von den jeweiligen Eigenschaften der Prüfsubstanz und der verwendeten Analysenmethode ab. Die Dämpfe sollten quantitativ so abgeschieden werden, daß eine anschließende Analyse möglich ist. Für manche Prüfsubstanzen werden sich mit Flüssigkeiten wie Hexan oder Ethylenglykol gefüllte Kühlfallen anbieten. Für andere wiederum mögen feste Adsorber zur Anwendung kommen.
Als Alternative zur Dampfabscheidung mit anschließender Analyse lassen sich online-Analysenmethoden wie z.B. die Chromatographie einsetzen, um die von einem bekannten Volumen an Trägergas mitgeführte Substanzmenge quantitativ zu bestimmen. Der Dampfdruck kann auch aus dem Masseverlust der eingesetzten Probe und bekanntem Trägergasvolumen bestimmt werden.
Wärmeaustauscher:
Zur Messung bei verschiedenen Temperaturen kann es notwendig sein, einen Wärmeaustauscher zur Temperierung des Trägergases in die Anordnung mit einzubauen.
Sättigungssäule:
Die Prüfsubstanz wird aus einer Lösung auf ein geeignetes inertes Trägermaterial aufgebracht. Das so beschichtete Trägermaterial wird in die Sättigungssäule eingebracht, die so dimensioniert und deren Gasdurchflußgeschwindigkeit so eingestellt werden sollte, daß eine vollständige Sättigung des Trägergases sichergestellt ist. Die Sättigungssäule muß thermostatisiert werden. Soll bei Temperaturen oberhalb der Raumtemperatur gemessen werden, müssen die Apparaturteile zwischen der Sättigungssäule und den Kühlfallen ebenfalls beheizt werden, um eine Kondensation der Prüfsubstanz zu vermeiden.
Um den durch Diffusion erfolgenden Massetransport zu reduzieren, kann im Anschluß an die Sättigungssäule ein Kapillarröhrchen angebracht werden (Abbildung 6b).
A.4. 1.6.6.2. Meßvorgang
Vorbereitung der Sättigungssäule:
Man löst die zu untersuchende Substanz in einem sehr flüchtigen Lösungsmittel und gibt sie einer ausreichenden Menge an Trägermaterial zu. Dabei ist eine genügende Menge an Prüfsubstanz hinzuzufügen, um die Sättigung für die gesamte Dauer des Tests zu gewährleisten. Das Lösungsmittel wird an der Luft oder im Rotationsverdampfer vollständig verdampft und das sorgfältig durchgemischte Material in die Sättigungssäule gefüllt. Nach dem Aufheizen der Probe im temperaturkontrollierten Bad wird trockener Stickstoff oder ein anderes geeignetes Trägergas durch die Apparatur geleitet.
Messung:
Man verbindet die Adsorptionsfallen oder den on-line-Detektor mit dem Ausgang der Säule und notiert die Zeit. Zu Beginn und in regelmäßigen Abständen während der Messungen kontrolliert man die Durchflußgeschwindigkeit mittels eines Blasenzählers (oder kontinuierlich mit einem Durchflußmesser).
Der Druck am Ausgang der Sättigungssäule muß gemessen werden. Dies geschieht entweder:
In Vorversuchen oder durch Schätzungen bestimmt man die erforderliche Zeit zur Abscheidung der für die verschiedenen Bestimmungsmethoden benötigten Substanzmenge. Als Alternative zur Dampfabscheidung mit anschließender Analyse lassen sich online-Analysenmethoden (z.B. die Chromatographie) einsetzen. Desgleichen sind Vorversuche zur Bestimmung der maximalen Durchflußgeschwindigkeit durchzuführen, bei der das Trägergas vollständig von dem Dampf der Substanz gesättigt wird, bevor der Dampfdruck bei einer gegebenen Temperatur berechnet wird. Zu diesem Zweck leitet man das Trägergas so langsam in die Sättigungssäule ein, daß sich für eine noch geringere Durchflußgeschwindigkeit kein größerer berechneter Dampfdruckwert ergibt.
Die verwendete Analysenmethode hängt von der Art der Prüfsubstanz ab (z.B. Gaschromatographie oder Gravimetrie).
Man bestimmt die Substanzmenge, die von einem bekannten Volumen an Trägergas mitgeführt wird.
A.4. 1.6.6.3. Bestimmung des Dampfdrucks
Der Dampfdruck berechnet sich aus der Dampfdichte W/V mit Hilfe folgender Gleichung:

Dabei bedeuten:
| p | = Dampfdruck (Pa) |
| W | = verdampfte Prüfsubstanz (g) |
| V | = Volumen des gesättigten Gases (m3) |
| R | = universelle molare Gaskonstante (J mol-1 K-1) |
| T | = Temperatur (K) |
| M | = Molargewicht der Prüfsubstanz (g/mol). |
Die gemessenen Volumina müssen wegen der herrschenden Druck- und Temperaturdifferenzen zwischen dem Durchflußmesser und den mit Thermostaten beheizten Sättigungssäulen entsprechend umgerechnet werden. Ist der Durchflußmesser den Adsorptionsfallen nachgeschaltet, so muß mit entsprechenden Korrekturen dem möglicherweise verdampften Inhalt der Fallen Rechnung getragen werden (1).
A.4 .2. Daten
Dampfdruckbestimmungen mit einer der vorstehend beschriebenen Methoden sollten bei mindestens zwei Temperaturen vorgenommen werden. Bevorzugt werden drei oder mehr Bestimmungen im Bereich von 0 °C bis 50 °C, um den Verlauf der Dampfdruckkurve zu überprüfen.
A.4. 3. Abschlußbericht
Im Prüfbericht ist, wenn möglich, folgendes anzugeben:
Wird eine Zustandsänderung (Phasenübergang, Zersetzung) festgestellt, sollten folgende Angaben notiert werden:
Alle zur Bewertung der Ergebnisse notwendigen Informationen und Bemerkungen sind zu notieren, insbesondere diejenigen über Verunreinigungen und den Aggregatzustand des Stoffes.
A.4. 4. Literatur
(1) OECD, Paris, 1981, Test Guideline 104, Decision of the Council C(81) 30 final.
(2) Ambrose, D. B. Le Neindre, B. Vodar, (Hrsg.): Experimental Thermodynamics, Butterworths, London, 1975, Vol. II.
(3) R. Weissberger (Hrsg.): Technique of organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Chapter IX, Interscience Publ. New York, 1959, vol. I, Part I.
(4) Knudsen, M.: Ann. Phys. Lpz., 1909, vol. 29, 1979; 1911, vol. 34, 593.
(5) NF T 20-048 AFNOR (Sept. 85). Chemical products for industrial use - Determination of vapour pressure of solids and liquids within range from 10 1 to 105 Pa - Static method.
(6) NF T 20-047 AFNOR (Sept. 85). Chemical products for industrial use - Determination of vapour pressure of solids and liquids within range from 10 3 to 1 Pa - Vapour pressure balance method.
(7) ASTM D 2879-86, Standard test method for vapour pressure-temperature relationship and initial decomposition temperature of liquids by isoteniscope.
(8) G. Messer, P. Röhl, G. Grosse und W. Jitschin. J. Vac. Sci. Technol., (A), 1987, vol. 5 (4), 2440.
(9) Ambrose, D.; Lawrenson, I.J.; Sprake, C.H.S. J. Chem. Thermodynamics 1975, vol. 7, 1173.
(10) B.F. Rordorf. Thermochimica Acta, 1985, vol. 85, 435.
(11) G. Comsa, J.K. Fremerey und B. Lindenau. J. Vac. Sci. Technol., 1980, vol. 17 (2), 642.
(12) G. Reich. J. Vac. Sci. Technol., 1982, vol. 20 (4), 1148.
(13) J.K. Fremerey. J. Vac. Sci. Technol., (A), 1985, vol. 3 (3), 1715.
Anlage 1
zu 67/548/EWG Anhang V A.4.
Schätzverfahren
Einleitung
Berechnete Dampfdruckwerte können verwendet werden:
Schätzverfahren
Der Dampfdruck von Flüssigkeiten und Feststoffen läßt sich durch Verwendung der modifizierten Watson-Korrelation abschätzen (a). Die einzige experimentelle Angabe, die benötigt wird, ist der Normal-Siedepunkt. Die Methode läßt sich über den Druckbereich von 105 Pa bis 10-5Pa einsetzen.
Ausführliche Angaben über die Methode sind dem "Handbook of Chemical Property Estimation Methods`(b) zu entnehmen.
Berechnungsverfahren
Nach (b) wird der Dampfdruck wie folgt berechnet:

Dabei bedeuten:
| T | = interessierende Temperatur |
| Tb | = Normal-Siedepunkt |
| Pvp | = Dampfdruck bei Temperatur T |
| Δ Hvb | = Verdampfungswarme |
| Δ Zb | = Kompressibilitatsfaktor (geschätzt auf 0,97) |
| m | = empirischer Faktor, abhängig vom Aggregatzustand bei der Temperatur T. |
Weiter gilt
 = KF (8,75 + R ln Tb)
= KF (8,75 + R ln Tb)
wobei KF ein empirischer Faktor ist, der die Polarität der Substanz berücksichtigt. In (b) sind die KF- Faktoren für verschiedene Arten von Substanzen aufgelistet.
Sehr häufig liegen Daten vor, bei denen ein Siedepunkt bei reduziertem Druck angegeben ist. In einem solchen Fall wird der Dampfdruck nach (b) wie folgt errechnet:

wobei T1 der Siedepunkt bei dem reduzierten Druck P1 ist.
1.6.7. Rotormethode (8, 11, 13)
1.6.7.1. Apparatur
Dieses Verfahren läßt sich mit einem Rotorviskositätsmeßgerät, wie in Abbildung 8 dargestellt, durchführen. Eine schematische Darstellung der Versuchsanordnung ist in Abbildung 7 enthalten.
Die typische Meßapparatur besteht aus einem in einem thermostatisierten Behälter angeordneten Sensorkopf (temperiert auf 0,1 °C). Auch das Probengefäß befindet sich in einem thermostatisierten Behälter (temperiert auf 0,01 °C). Um eine Kondensation zu vermeiden, werden alle anderen Teile der Anordnung auf einer höheren Temperatur gehalten. Eine Hochvakuum-Pumpe ist über Hochvakuum-Ventile mit dem System verbunden.
Der Sensorkopf besteht aus einer in einem Rohr angeordneten Stahlkugel (Durchmesser 4 bis 5 mm), die in einem Magnetfeld stabilisiert wird und normalerweise mit einer Kombination aus Permanentmagneten und Steuerspulen arbeitet.
Die Kugel wird durch über die Spulen erzeugte Drehfelder zum Rotieren gebracht. Mit Hilfe von Aufnahmespulen, die die stets vorhandene geringe laterale Magnetisierung der Kugel messen, läßt sich deren Drehgeschwindigkeit bestimmen.
1.6.7.2. Meßvorgang
Wenn die Kugel eine vorgegebene Drehgeschwindigkeit v(0) (i.allg. etwa 400 U/s) erreicht hat, wird die weitere Energiezufuhr gestoppt, und es kommt auf Grund der Gasreibung zu einer Abbremsung.
Der Rückgang der Drehgeschwindigkeit wird in Abhängigkeit von der Zeit gemessen. Da die durch das Magnetfeld erzeugte Reibung im Vergleich zur Gasreibung vernachlässigbar ist, wird der Gasdruck p wie folgt angegeben:

Dabei bedeuten:
 |
= durchschnittliche Geschwindigkeit der Gasmoleküle |
| r | = Radius der Kugel |
| ρ | = Massendichte der Kugel |
| σ | = Koeffizient der tangentialen Momentübertragung (σ = 1 für eine Kugel mit idealer Sphäre) |
| t | = Zeit |
| v(t) | = Drehgeschwindigkeit nach der Zeit t |
| v(0) | = Anfangsdrehgeschwindigkeit. |
Diese Gleichung kann auch wie folgt geschrieben werden:
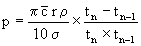
wobei tn, tn-1 die für eine bestimmte Anzahl N von Umdrehungen erforderlichen Zeiten sind. Diese Zeitintervalle tn und tn-1 folgen aufeinander, und tn > tn-1.
Die durchschnittliche Geschwindigkeit des Gasmoleküls wird durch folgende Gleichung angegeben:

Dabei bedeuten:
| T | = Temperatur |
| R | = universelle molare Gaskonstante |
| M | = Molargewicht. |
Abschlußbericht
Bei Anwendung des Abschätzverfahrens ist mit dem Bericht eine ausführliche Dokumentation zur Art der Berechnung beizufügen.
Literatur
(a) K.M. Watson, Ind. Eng. Chem., 1943, vol. 35, 398
(b) W.J. Lyman. W.F. Reehl, D.H. Rosenblatt. Handbook of Chemical Property Estimation Methods, Mc Graw-Hill, 1982.
Anlage 2
zur RL 67/548/EWG Anhang V A.2
Abbildungen
Abbildung 1: Apparatur zur Bestimmung der Dampfdruckkurve nach der dynamischen Methode
Abbildung 2a: Apparatur zur Bestimmung der Dampfdruckkurve nach der statischen Methode (unter Verwendung eines U-Rohr-Manometers)
Abbildung 2b: Apparatur zur Bestimmung der Dampfdruckkurve nach der statischen Methode (unter Verwendung eines Druckaufnehmers)
Abbildung 3: Isoteniskop (siehe Literaturstelle 2)
Abbildung 4: Apparatur zur Bestimmung der Dampfdruckkurve mit der Dampfdruckwaage
Abbildung 5: Beispiel einer Apparatur zur Verdampfung bei niedrigem Druck nach der Effusionsmethode - Effusionszellvolumen: 8 cm3
Abbildung 6a: Beispiel für ein Durchflußsystem zur Bestimmung des Dampfdrucks mit der Gassättigungsmethode
Abbildung 6b: Beispiel für ein System zur Bestimmung des Dampfdrucks mit der Gassättigungsmethode - unter Verwendung eines der Sättigungskammer nachgeschalteten Kapillarröhrchens
Abbildung 7: Beispiel für eine Versuchsanordnung mit dem Hochgeschwindigkeitsrotor; Apparatur zur Dampfdruckmessung
Abbildung 8: Beispiel für einen Hochgeschwindikeitsrotor-Meßkopf
| |
weiter . | |
(Stand: 04.08.2022)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: ab 105.- € netto
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}